概述
本型又名山菲利普(Sanfilippo)综合征,为常染色体隐性遗传性病。该型的临床特征和生化异常均不同于Ⅰ、Ⅱ两型,其特征为反应迟钝程度很严重,但周身病变比较轻微。与Ⅰ型不同之处为容貌不似承霤病,侏儒也不明显,亦无角膜浑浊或心脏病等并发症。肘、膝关节有轻度挛缩。即使有肝脾肿大,也很轻微。该症最早由Sanfilippo(1963)报道,以后Mekusiek将其分类为黏多糖贮积症Ⅲ型或称Sanfilippo综合征。
病因
病因为常染色体隐性遗传。
发病机制
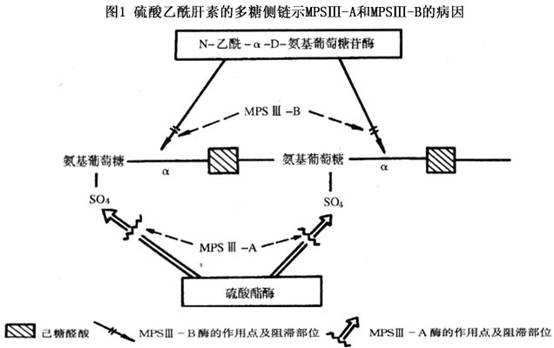
根据成纤维细胞混合培养结果,可将本病分为A、B、C、D四型。A型是由于溶酶体乙酰肝素-N-硫酸酯酶缺乏引起;B型是由于N-乙酰-α-D-氨基葡萄糖苷酶(图1)引起;C型是由于乙酰CoA-α-葡萄糖胺-N-乙酰转移酶缺乏引起;D型是由于N-乙酰-α-D-氨基葡萄糖苷-6-硫酸酯酶缺乏引起。上述酶缺乏使硫酸乙酰肝素不能逐步降解,而在肝及组织内贮积,并大量随尿排出。虽然酶的缺陷不同,但4型的症状完全相同,临床上不能区分。4型细胞可以相互矫正代谢缺陷,使4型病人细胞内的黏多糖沉积都得到矫正。这种“交叉矫正”现象大概是因为不同型病人的成纤维细胞可以相互供给细胞缺乏的酶所致。 病理:病理...[详细]
 病理:病理检查可见肝细胞和库普弗细胞以及直肠黏膜细胞中有大空泡形成。脑室扩大,脑实质神经元严重变性和脱失,而残留有神经元气球样变,内含脂质而外观肿胀;脑灰质鞘脂大量增多。基底神经节和视神经节细胞变性肿胀。脑的这些病变就是临床上严重智力低下的原因。 电镜检查显示有不同类型的细胞质包涵体;具有均匀颗粒物质及小同心圆性膜小体空泡,具有亲水外膜和斑马小体空泡。 组织化学检查证明脑、肝、肾所沉积的黏多糖主要为硫酸乙酰肝素,脑组织中还有糖脂、GM神经节苷脂含量增加。
病理:病理检查可见肝细胞和库普弗细胞以及直肠黏膜细胞中有大空泡形成。脑室扩大,脑实质神经元严重变性和脱失,而残留有神经元气球样变,内含脂质而外观肿胀;脑灰质鞘脂大量增多。基底神经节和视神经节细胞变性肿胀。脑的这些病变就是临床上严重智力低下的原因。 电镜检查显示有不同类型的细胞质包涵体;具有均匀颗粒物质及小同心圆性膜小体空泡,具有亲水外膜和斑马小体空泡。 组织化学检查证明脑、肝、肾所沉积的黏多糖主要为硫酸乙酰肝素,脑组织中还有糖脂、GM神经节苷脂含量增加。临床表现
进行性智力低下是本症的最显著特征。生后1岁内智力发育均正常或仅有轻度落后,智力低下一般在4~7岁出现,10岁时已很严重。在智力低下进行性加重的同时,可出现进行性神经症状,如抽风、运动过多、痉挛性四肢瘫、全身无力、攻击性行为等,此为该症最突出的症状。 身材改变较轻或表现正常,仅有1/4的病人表现矮小。大多数病人面部表现正常,少数病人表现头大、面容丑陋、腹部膨隆、进行性耳聋、关节僵直和手屈曲呈爪状。骨骼改变仅有多发性成骨不全和顶骨后部致密、增厚。这些改变对诊断本病具有一定程度的特异性。肝、脾肿大为轻度到中度,无角膜浑浊。无心脏受累。
并发症
本病可并发,痉挛性四肢瘫、攻击性行为等。即使有肝脾肿大,也很轻微。
实验室检查
尿中排泄的大量黏多糖是硫酸乙酰肝素,MPSⅢ-B病人血清中酶的活性为正常者的2%~16%,而杂合子的酶活性在正常者的35%以下。血液中淋巴细胞、中性粒细胞和骨髓细胞可发现有异染性包涵体。
其他辅助检查
X线检查:X线表现与黏多糖贮积症Ⅰ、Ⅱ型相似,约半数病人可出现Ⅰ型改变,但程度较轻,其中仅1/4的病人发生侏儒。另外半数病人可出现长骨和肋骨增大畸形。部分病人腰椎椎体呈双凸形,顶骨后部密度增高、增厚。
诊断
根据临床特征和尿液分析结果,应怀疑该型综合征的诊断,而确诊必须依靠证实有特异的酶缺陷,因为本型4个亚型临床表现相似。
治疗
同黏多糖贮积症Ⅰ型。加强出生前诊断为本症的惟一预防措施。
预后
可能在儿童期死亡,也可存活较长,多死于呼吸道感染。