概述
扭转痉挛(torsion spasm)又称特发性扭转痉挛(idiopathic torsion spasm,ITS)、扭转性肌张力障碍(torsion dystonia)、原发性肌张力障碍(primary dystonia),临床以肌张力障碍及四肢、躯干以至全身剧烈不随意扭转为特征。分为原发性及继发性,原发性多见。 扭转痉挛是全身性肌张力障碍中的一种临床表现类型,也是原发性肌张力障碍中最有特征性的表现。以年轻人发病多见,病初只表现局限性的肌张力障碍症状,以后波及全身,造成扭转痉挛。可有阳性家族史,尤其在Ashkenazic犹太人中。
病因
部分常染色体显性遗传患者是9号染色体长臂(9q34)DYT1基因突变所致。由于多巴反应性肌张力障碍造成的扭转痉挛,则基因位于常染色体14q。特发性扭转痉挛病因不明,多为散发,少数有家族遗传史。有许多研究表明,约85%的扭转痉挛患者存在较低外显率的常染色体显性遗传基因,少部分人有新的突变。在一般人中是否存在常染色体隐性遗传或X性连锁遗传类型尚不能肯定。在第一代遗传家族的亲属中,疾病的危险性约有20%。 症状性扭转痉挛见于累及基底核的各种疾病,如感染(脑炎后),变性(肝豆状核变性,Hallervorden-Spatz病),中毒(特别是CO及左旋多巴,酚噻嗪或丁酰苯类过量),代谢障碍(基底...[详细]
发病机制
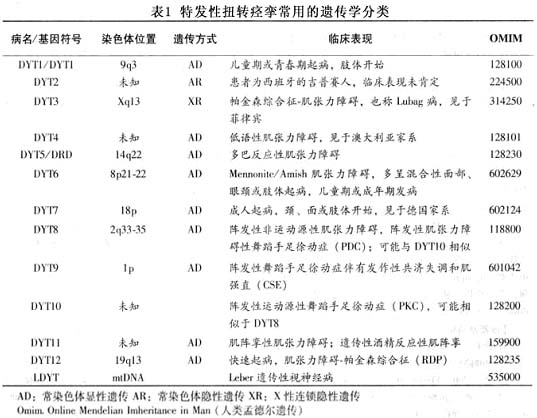
Eldridge等(1970)发现两种遗传类型。一种是常染色体显性遗传,另一种是常染色体隐性遗传,Zeman等(1978)报道偶尔也有配子细胞突变引起者。 近年来分子遗传学研究已发现9个位点基因突变可导致ITS,目前国际最常用的遗传学分类见表1。 继发性扭转痉挛的临床表现酷似原发性,故推想原发性扭转痉挛也是基底核的一种疾病。其发病机制可能是调节肌张力的反馈机制紊乱,出现临床上奇怪的不自主姿势、体位和动作,甚至有自发性的肌肉痉挛。 原发性肌张力障碍的病理改变一直没有肯定的结论。遗传型患者脑组织光镜检查未发现纹状体及苍白球有明显异常,显性遗传型患者血浆内多...[详细]
 继发性扭转痉挛的临床表现酷似原发性,故推想原发性扭转痉挛也是基底核的一种疾病。其发病机制可能是调节肌张力的反馈机制紊乱,出现临床上奇怪的不自主姿势、体位和动作,甚至有自发性的肌肉痉挛。 原发性肌张力障碍的病理改变一直没有肯定的结论。遗传型患者脑组织光镜检查未发现纹状体及苍白球有明显异常,显性遗传型患者血浆内多巴胺-β-羟化酶及去甲肾上腺素含量增多,意义不明。 症状性扭转痉挛的基底核病变视病因不同而病理各异。在有特异性病理形态变化的继发性(症状性)扭转痉挛,大多伴有上脑干附近与运动有关的中枢神经损害。从继发性(症状性)扭转痉挛的肯定病理改变来看,肌张力障碍的病变主要在新纹状体、苍白球、丘脑和(或)它们的联络纤维。已发现非特异性的病理改变包括壳核、丘脑及尾核的小神经元变性消失,基底核的脂质及脂色素增多。
继发性扭转痉挛的临床表现酷似原发性,故推想原发性扭转痉挛也是基底核的一种疾病。其发病机制可能是调节肌张力的反馈机制紊乱,出现临床上奇怪的不自主姿势、体位和动作,甚至有自发性的肌肉痉挛。 原发性肌张力障碍的病理改变一直没有肯定的结论。遗传型患者脑组织光镜检查未发现纹状体及苍白球有明显异常,显性遗传型患者血浆内多巴胺-β-羟化酶及去甲肾上腺素含量增多,意义不明。 症状性扭转痉挛的基底核病变视病因不同而病理各异。在有特异性病理形态变化的继发性(症状性)扭转痉挛,大多伴有上脑干附近与运动有关的中枢神经损害。从继发性(症状性)扭转痉挛的肯定病理改变来看,肌张力障碍的病变主要在新纹状体、苍白球、丘脑和(或)它们的联络纤维。已发现非特异性的病理改变包括壳核、丘脑及尾核的小神经元变性消失,基底核的脂质及脂色素增多。临床表现
1.各年龄均可发病,常染色体隐性遗传常在儿童期起病(儿童期肌张力障碍),多有家族史。散发病例及常染色体显性遗传起病年龄较迟,外显率多不完全。成年期起病(成年期肌张力障碍)多为散发,常可找到继发病因,约20%的患者最终发展为全身性肌张力障碍。 2.典型症状 常从一侧或两侧下肢开始,足呈内翻跖曲(图1),行走时足跟不能着地,随后躯干及四肢发生不自主扭转运动和姿势异常,以躯干为轴扭转或螺旋样运动最具特征性(图2),动作多变无规律,自主运动或精神紧张时扭转痉挛加重,睡眠时消失。颈肌受累出现痉挛性斜颈,面肌受累出现不自主挤眉弄眼、眼睑痉挛、张口闭口、牵嘴歪舌、舌伸扭动等怪异表情(口下颌肌张力...[详细]

 3.常染色体显性遗传家族成员中,可有多人患病或多种顿挫型局限性症状,如眼睑痉挛、斜颈、书写痉挛和脊柱侧弯等,多自上肢开始,长期局限于起病部位,即使进展为全身型,症状亦较轻微。极少见情况下,某些不明原因的扭转痉挛可迅速进展,临床症状急骤恶化,最终导致死亡。Vamonde等(1994)报道2例儿童期起病,表现全身性扭转痉挛,迅速进展,全身症状明显,很快死亡,称为肌张力障碍风暴(dystonic storms)。
3.常染色体显性遗传家族成员中,可有多人患病或多种顿挫型局限性症状,如眼睑痉挛、斜颈、书写痉挛和脊柱侧弯等,多自上肢开始,长期局限于起病部位,即使进展为全身型,症状亦较轻微。极少见情况下,某些不明原因的扭转痉挛可迅速进展,临床症状急骤恶化,最终导致死亡。Vamonde等(1994)报道2例儿童期起病,表现全身性扭转痉挛,迅速进展,全身症状明显,很快死亡,称为肌张力障碍风暴(dystonic storms)。并发症
部分患者可能有智能减退。因不自主运动而不能从事正常的活动,晚期病例可因骨骼畸形、肌肉挛缩而发生严重残废。
实验室检查
血电解质、药物、微量元素及生化检查,有助于鉴别诊断及分类。
其他辅助检查
1.CT、MRI检查,正电子发射断层扫描(PET)或单光子发射断层扫描(SPECT),对鉴别诊断有意义。 2.基因分析 对确诊某些遗传性肌张力障碍疾病有重要意义。
诊断
扭转痉挛的诊断并不困难,因颈部、躯干四肢及骨盆等奇异的扭转运动为本病所特有,可以一目了然。
治疗
1.本病尚无有效疗法,严重特发性扭转痉挛可施行立体定向丘脑腹外侧核后半部毁损术,常可复发。症状性扭转痉挛可对因治疗,药物诱发须立即停药,选用可缓解肌张力障碍药物。①镇静剂地西泮(安定)2.5~5mg或硝西泮(硝基安定)5~7.5mg,3次/d,对部分病例有效;②左旋多巴疗效仍有争议,少数报道认为有效,左旋多巴500~1000mg/d,对变异型特发性扭转痉挛(多巴反应性肌张力障碍)有戏剧性效果;有人认为左旋多巴使症状加重;③大剂量抗乙酰胆碱药治疗特发性扭转痉挛有效,苯海索(benzhexol)20mg/d,分2~3次口服,儿童有效;苯海索(三己芬迪)20~80mg/d,分3~4次口服;④抗精神病...[详细]
预后
原发性扭转痉挛的转归差异较大,起病年龄和部位是影响预后的两个主要因素。起病年龄早(15岁以前)以及自下肢起病者,大多不断进展,最后几乎都发展为全身型,预后不良,多在起病若干年后死亡,自行缓解甚少。成年期起病且症状自上肢开始者预后较好,不自主运动趋向于长期局限于起病部位。常染色体显性遗传型或分散型的预后较隐性遗传型好,因为前者起病年龄晚且多自上肢起病。
预防
有遗传背景的患者,预防显得更为重要。预防措施包括避免近亲结婚,推行遗传咨询、携带者基因检测及产前诊断和选择性人工流产等,防止患儿出生。早期诊断、早期治疗、加强临床护理,对改善患者的生活质量有重要意义。