概述
组织细胞增多病X(histiocytosis X)是由大量增生的染色淡而大的组织细胞在眼眶或全身各脏器、软组织和骨中堆积引起的病变。根据累及的部位不同又分为局部单灶性和全身多灶性。年龄越小越容易发展成为多灶性病灶,患者年龄偏大,单灶性病变可能性大。1~2岁内多系统如皮肤、内脏、淋巴结和眼眶受侵犯者视为累-赛病(Letterer-Siwe disease);年龄稍大的小儿表现为典型的三联征:颅骨缺损,突眼和尿崩症称为汉德-许勒尔-克思斯琴病(Hand-Schüller-Christian disease);单一性病变叫嗜酸性细胞肉芽肿(eosinophilic granuloma)。众多学者对该...[详细]
病因
发病机制不清,可能是一种免疫异常性疾病,特别与T淋巴细胞异常有关,单克隆抗体OKT-6可使Langerhans细胞表面膜受体染色。
发病机制
机制尚不明确。
临床表现
孤立的骨病变多见于颅骨,依次为骨盆、脊柱、肋骨和四肢长骨。嗜酸性肉芽肿也可以是多灶性的。颅骨的额骨和顶骨是常见的病变区,当眶骨受累时外颞上眶缘是最普遍的部位(图1),偶尔眶骨和颅骨广泛受累。病变区肿胀,有压痛或疼痛,骨肿胀,骨右眼眶颞上方肿块状新生物,上睑肿胀、下垂。眼球前突、向下移位。病变发生在骨的板障层,突破眶骨膜,引起上睑外侧软组织炎症和泪腺发炎,故临床类似于眼眶的皮样囊肿和泪腺炎的表现。极个别病变可累及角膜、巩膜和葡萄膜。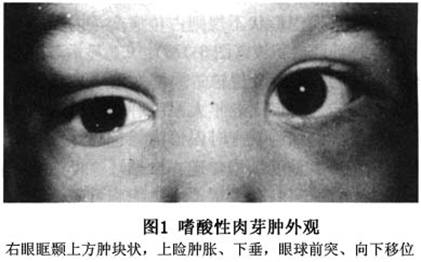
并发症
肉芽肿侵及角膜、巩膜或葡萄膜时可出现相应的不同症状。
实验室检查
1.免疫学检查 可能发现部分指标异常。 2.病理学检查 活检标本的组织病理学显示最明显的细胞是大而淡染的组织细胞,核位于细胞中心,呈卵圆形,空泡状,核膜轻度凹陷,嗜酸性粒细胞质内有较多的颗粒。在组织细胞浸润的背景中,可见不同数量的嗜酸性粒细胞、淋巴细胞、浆细胞和多核巨细胞(图2)。病灶区内基质少,但血管多,易出血,故可见含铁血黄色素的巨噬细胞。旧病变中的单核细胞和多核细胞发生脂化,骨内的一些病变可以自愈,病变区纤维化而呈现硬化灶。 电子显微镜证实单核细胞组织细胞质内有特殊的颗粒呈棒状,中心有条纹,末端有开口的空泡,酷似球拍,故叫球拍小体,又名Birbeck颗粒...[详细]
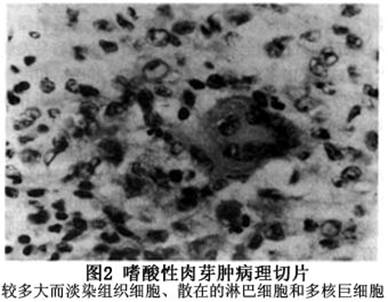 电子显微镜证实单核细胞组织细胞质内有特殊的颗粒呈棒状,中心有条纹,末端有开口的空泡,酷似球拍,故叫球拍小体,又名Birbeck颗粒或Langerhans颗粒。多核巨细胞含有丰富的线粒体、滑面内质网,但无Langerhans颗粒。所以Langerhans颗粒是树突状组织细胞(Langerhans细胞)的标记。现已知嗜酸性细胞肉芽肿是Langerhans细胞的增生,该细胞常存在皮肤的表皮层,这就可解释嗜酸性细胞肉芽肿的组织细胞高度嗜表皮的原因。
电子显微镜证实单核细胞组织细胞质内有特殊的颗粒呈棒状,中心有条纹,末端有开口的空泡,酷似球拍,故叫球拍小体,又名Birbeck颗粒或Langerhans颗粒。多核巨细胞含有丰富的线粒体、滑面内质网,但无Langerhans颗粒。所以Langerhans颗粒是树突状组织细胞(Langerhans细胞)的标记。现已知嗜酸性细胞肉芽肿是Langerhans细胞的增生,该细胞常存在皮肤的表皮层,这就可解释嗜酸性细胞肉芽肿的组织细胞高度嗜表皮的原因。其他辅助检查
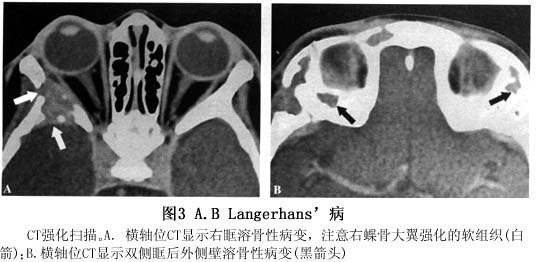
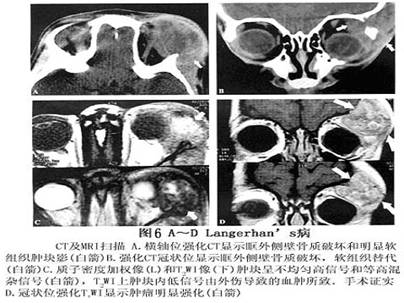
1.X线检查 颅骨是最好发部位,常侵及多块颅骨,尤以颅盖骨为甚。病灶大小不等,常相互融合呈大块“地图样”骨缺损,这种表现颇有特征。颅骨表现多从板障开始,随病灶的扩展,使内外骨板破坏,局部软组织隆起肿胀,骨质破坏区的边缘锐利,通常无硬化,也无骨膜反应是其特点。眶窝骨质破坏以累及眼眶之外上缘为多,并出现同侧突眼。 2.超声波探查 病变累及眼眶时,眼眶内可探及异常回声区,边缘不清,形态不规则,内回声强弱分布不均匀,声衰减明显,后界显示较弱或不清,缺乏可压缩性。 3.CT扫描 眶骨骨质破坏呈溶骨性,边缘清晰,局部软组织肿胀,可以侵犯眼外肌、泪腺、甚至眼球,颞肌亦可侵犯,增强后...[详细]
 4.MRI 病变常位于肌锥外,表现为不规则肿块,病变发展到一定程度可破坏眶壁,侵及颞窝或颅腔。侵及颅内者,类似其他颅眶沟通瘤表现,但境界多较清楚,T1加权像呈中低信号,T2加权像呈中高信号。肿块侵及眼外肌致其受压、移位,境界不清,眼球常有突出(图5,6)。
4.MRI 病变常位于肌锥外,表现为不规则肿块,病变发展到一定程度可破坏眶壁,侵及颞窝或颅腔。侵及颅内者,类似其他颅眶沟通瘤表现,但境界多较清楚,T1加权像呈中低信号,T2加权像呈中高信号。肿块侵及眼外肌致其受压、移位,境界不清,眼球常有突出(图5,6)。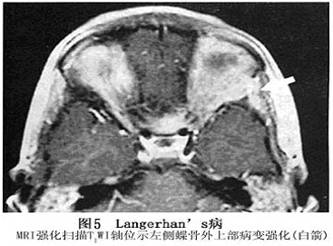
诊断
患者常为儿童,在颞上外眶缘处扪及肿块,有压痛,就应想到嗜酸性肉芽肿的可能性。X线照片显示不规则、锯齿状的溶骨区,无硬化边界。CT检查除发现溶骨性缺损外,还发现外上方软组织密度增加,局部高起。对这样的病变应做活体组织检查,发现大量的组织细胞增生,电子显微镜见细胞质内有Langerhans颗粒,对年龄较大的患者可诊断嗜酸性肉芽肿。
治疗
眼眶嗜酸性肉芽肿为单灶性病变,手术刮除能消除病灶,皮质类固醇内注射能治愈病灶,但注射时切勿穿过硬脑膜,将药物注射到颅内。有作者主张在病灶刮除后,给局部小剂量分次放射治疗,总剂量6~8Gy,效果更好。也可以观察,因病变为良性,且有自愈倾向。对复发病例要全身用皮质类固醇和抗代谢药物治疗,Song等报道1例9岁男孩眼眶外上方嗜酸性肉芽肿,刮除术后6个月,MRI检查发现眶内肿块复发,用泼尼松和长春新碱治疗,效果显著。
预后
本病的预后与发病时的年龄、病变程度、是否伴有器官功能障碍等情况有关。发病年龄越早预后越差。本病的部分病例可以自行缓解。治疗可根据病情轻重采用保守疗法、激素疗法和化学疗法,亦可考虑小剂量放射治疗。
预防
目前没有相关内容描述。